澳大利亚拥有一个成熟且规模庞大的公共和私人医疗保健系统,对医疗器械的需求也十分旺盛。然而,想要在澳大利亚销售、进口、出口医疗器械,必须按照相关规定完成注册审核并获得准入证书。这篇文章将详细介绍这个过程。
一、澳大利亚的监管法规
早在1966年,澳大利亚就通过了《医疗用品法案》,对医疗用品进行管理。现在主要的管理法案是1989年的《医疗用品法案》,以及2002年颁布的《医疗器械法规》。这两部法规的执行和监督由澳大利亚联邦健康与老年护理部的下属机构TGA负责。因此,想要向澳大利亚销售、进出口医疗器械的制造商需向TGA提交市场准入申请。
二、澳大利亚医疗器械的定义和分类
澳大利亚将医疗器械定义为由工具、仪器、用具或其他物品以及必要的附件或软件组成的治疗品。这些治疗品主要通过非药理学、免疫学或代谢方式达到主要作用,尽管这些方式可能辅助其作用。根据风险等级,医疗器械被分为Ⅰ级、Ⅱa级、Ⅱb级、Ⅲ级、以及AIMD级(活性植入医疗器械)。
三、澳大利亚医疗器械的市场准入
TGA将医疗器械分为三类进行管理:豁免、备案和注册。每种类型的医疗器械在上市销售前都必须获得政府的批准,并且符合医疗器械的基本要求。高风险的医疗器械必须通过TGA的评估并在上市前获得批准。而低风险的医疗器械可以由企业自行评估,只要符合质量和安全条件就可以进入市场。所有在澳大利亚生产的医疗器械必须符合GMP标准,并在洁净和无污染的环境下生产。
四、澳大利亚医疗器械上市流程解析
医疗器械在澳大利亚的上市流程是一项严谨而繁琐的工作,需要满足多个关键角色的职责,明确医疗器械的定义及风险等级,以及完成符合性评估和ARTG注册。
(1)明确关键角色
首先,医疗器械在澳大利亚的注册上市过程涉及两个关键角色,即制造商(Manufacturer)和保荐人(Sponsor)。制造商负责医疗器械的设计、生产、包装、发货等,而保荐人是一位澳大利亚本地公民或公司,负责向TGA(治疗性商品管理局)申请Sponsor注册号,以帮助完成注册过程。
(2)明确医疗器械的定义及风险等级
接下来,需要明确医疗器械的定义及风险等级。1989年的《医疗用品法案》对医疗器械的定义及要点如下:

在澳大利亚,符合《医疗用品法案1989》定义的医疗器械需要向TGA提交注册申请。根据风险程度和预期用途的不同,医疗器械被分为五类风险等级,风险级别越高,TGA监管越严格。

(3)申请符合性评估证明
在医疗器械注册上市之前,制造商需要实施合格的符合性评估程序,并接受TGA的审核,获得有效的符合性评估证明。TGA认可的符合性评估证明包括TGA Conformity Assessment Evidence、EC Certificate,以及基于EC-MRA和EFTA-MRA颁发的证书。
如果选择TGA符合性评估证据,证书申请过程如下:
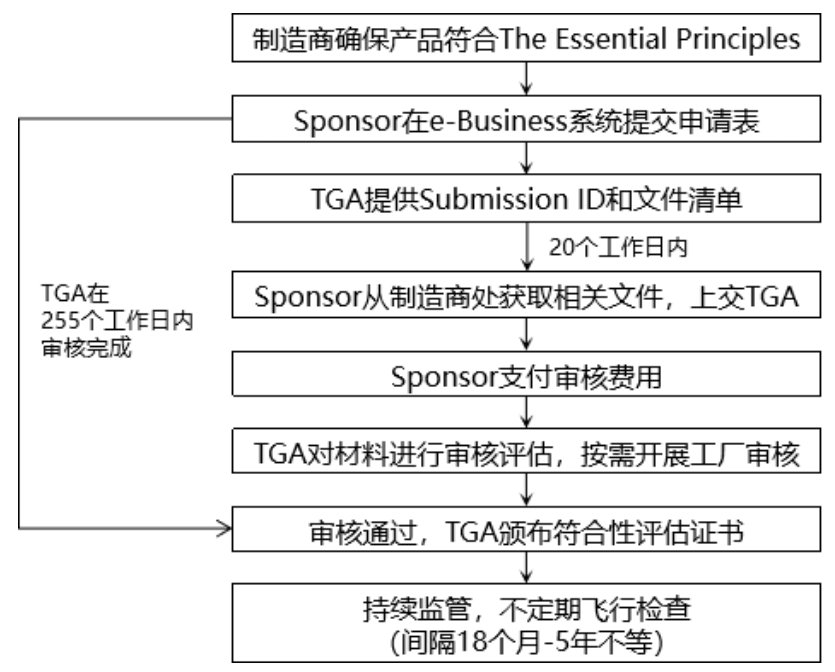
(4)提交ARTG注册
完成符合性评估后,制造商和Sponsor需要提交ARTG注册,获得单个医疗器械的准入证书,以完成产品列示。只有当医疗器械被列入澳大利亚治疗性商品登记处(ARTG)后,才能在澳大利亚市场上销售。
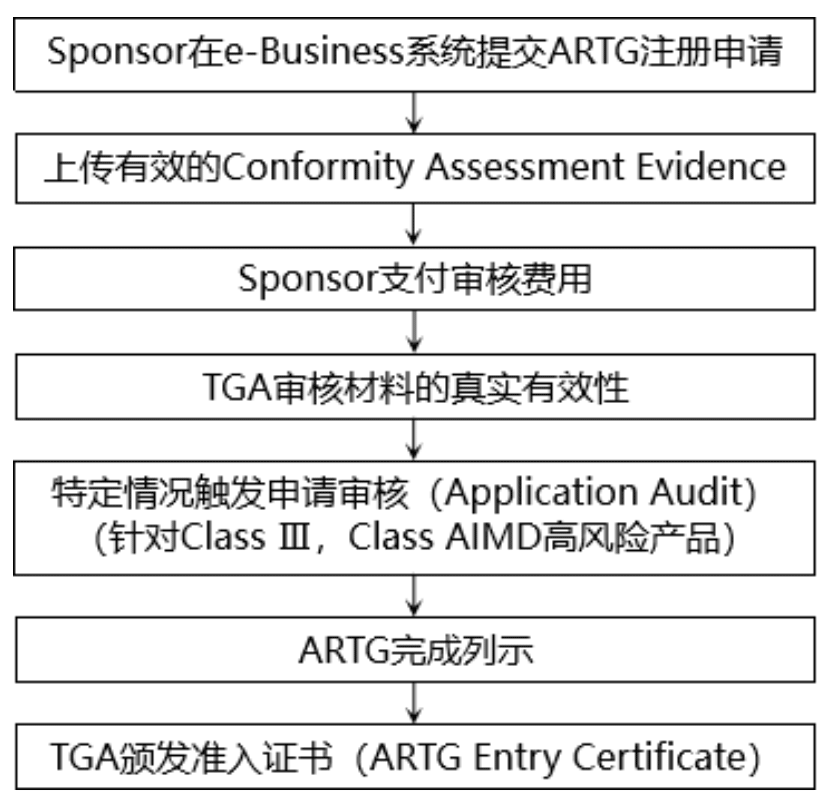
五、澳大利亚医疗器械的质量体系
政府规定所有医疗器械生产企业的生产过程都必须符合与其生产的器械相关的质量要求,并具备质量保证的手段和程序。为了实现这一目标,澳大利亚执行了良好制造实践(GMP)制度,同时也向ISO 9000质量体系标准靠拢。2017年起,澳大利亚开始实施医疗器械单一审核程序,并接受有资格的检查机构提供的MDSAP认证证书。
六、澳大利亚医疗器械产品的市场后管理
这主要通过上市后警戒管理体系来实现。澳大利亚通过不良事件的调查报告、上市产品的实验室检验和监测的活动,确保所有上市后的医疗器械符合法规的规定。其上市后不良事件监测的规定非常成熟,程序详尽,结合了原则性和灵活性,具有强大的可操作性。
七、澳大利亚医疗器械产品的注册变更规定
产品在获得准入上市后,其信息和状态仍需持续被监管。如果产品出现实质性变更,制造商和Sponsor必须及时通知治疗性商品管理局(TGA)。Sponsor需要登录e-Business系统提交注册变更申请。这些变更可能包括制造商、保荐人、产品的信息的变更,符合性评估证明信息的变更,产品预期用途、临床适应症的变更,以及产品所属种类、GMDN代码的变更。
总的来说,要在澳大利亚销售、进口或出口医疗器械,必须通过一系列严格的准入和注册过程。这些过程保证了医疗器械的质量、安全性和有效性,进而保障了医疗保健系统的质量。


© 2025. All Rights Reserved.沪ICP备2023007705号-2 沪公网安备31011502009912号
沪公网安备31011502009912号